Simulate under a HiSSE model
simulateHisse.RdFlexible simulation function allowing checkpointing
Arguments
- turnover.rates
a vector of turnover rates.
- eps.values
a vector of extinction fractions.
- transition.rates
a matrix of transition rates.
- max.taxa
have the simulation stop at
max.taxasurviving taxa.- max.t
have the simulation stop at
max.theight (including stem).- max.wall.time
have the simulation stop at this many seconds of running.
- x0
the starting state.
- nstart
how many taxa you want to start from. Usual values are 1 (start with single lineage) or 2 (crown group).
- checkpoint.file
if you want to save progress (so you can restart from last saved point) include the name of a file.
- checkpoint.frequency
if there is a checkpoint file, frequency (in terms of number of steps: births, deaths, or transitions) at which this is saved.
- checkpoint.start.object
if you are starting from a checkpointed.file, the object loaded from that file. Typically called
checkpoint.result.- override.safeties
simulate even if there is no limit on number of taxa, depth of tree, or wall time.
- mass.extinction.heights
vector of times above the root [NOT from the present] to have a mass extinction.
- mass.extinction.magnitudes
vector of per species extinction probability at mass extinctions.
Details
Note that currently, the simulator assumes turnover.rates, eps.values, and transition rates are in the same state order. Remember that turnover.rates are birth + death rates, and eps.values are death / birth rates.
We strongly advise putting some limits to stop the run. These can be any combination of max.taxa, max.t, and max.wall.time. For example, you could set up the run to stop when you hit 1000 taxa or 3600 seconds (one hour) of running on your computer, whichever comes first. If you want to run with no limits, you have to specify override.safeties=TRUE, and you should know what you're doing here (the runs might never finish). There is only one starting state (x0), which should be the index into the rate vectors, starting with 0: that is, if your states are 0A, 0B, 1A, and 1B, x0=0 would start in 0A, x0=3 would start in 1B. nstart lets you choose how many taxa to start with: it could be one lineage to start from a single taxon, in which case you'll have to wait some time for the first speciation event, or you could start with two, to start simulation with a clade of this size (though, if extinction is nonzero, you are not guaranteed to have the final crown group include both of these descendants). You can choose numbers higher than two, to start with, say, a 50-species polytomy. We're not sure why you would. You can add checkpointing: runs take a long time, and if it crashes, this will let you start from the same point in the last saved file (though with a different seed, unless you specify this yourself). If you want to save checkpoints as you go along, specify a file in checkpoint.file. Every checkpoint.frequency events, where an event is a speciation, extinction, or transition event, it will save current progress to a file. The smaller this value, the more frequently the simulation will be saved: fewer lost steps if you have to re-simulate, but slower during the run because it spends time writing to disk. If you want to start from a checkpointed analysis, load the file into R and then specify the object within R [not the file] containing the checkpointed results (by default, saved in an object called checkpoint.result) and the simulation will continue from that point.
Results are returned in a list, containing a dataframe with the outcome of the simulation (one row per edge in the tree, and containing information on the lengths, ancestor, tipward height from the original root, and state at the tipward end of the edge (you can get state at the rootward end by getting the tipward state of the ancestral edge). It may be of interest to know how many events of each type occurred, which is saved in $birth.counts, $death.counts, $transition.counts. The total number of surviving taxa is stored in $n.surviving.
The output can be returned as an ape phylo tree by passing the $results element of the final output to SimToPhylo().
Value
This returns a list, with the following elements:
- $results
a dataframe containing the simulation output: states at nodes and tips, ancestor-descendant pairs, branch lengths.
- $birth.counts
a vector, in the same state order as turnover.rates et al., that includes the counts for the number of times species in each state speciated.
- $death.counts
same as birth.counts, but counting extinction events in each state outside of mass extinctions.
- $mass.extinction.death.counts
total number of species that went extinct during a mass extinction event (which is assumed to be trait-independent).
- $transition.counts
matrix of counts of transitions from one state to another.
- $n.surviving
the number of taxa alive at the end of the simulation.
References
Beaulieu, J.M, and B.C. O'Meara. 2016. Detecting hidden diversification shifts in models of trait-dependent speciation and extinction. Syst. Biol. 65:583-601.
Examples
# \donttest{
simulated.result <- SimulateHisse(c(.3, .1), c(.1, 0),
matrix(c(NA, 0.2, .3, NA), nrow=2), max.taxa=35, x0=1)
par(mfcol=c(1,2))
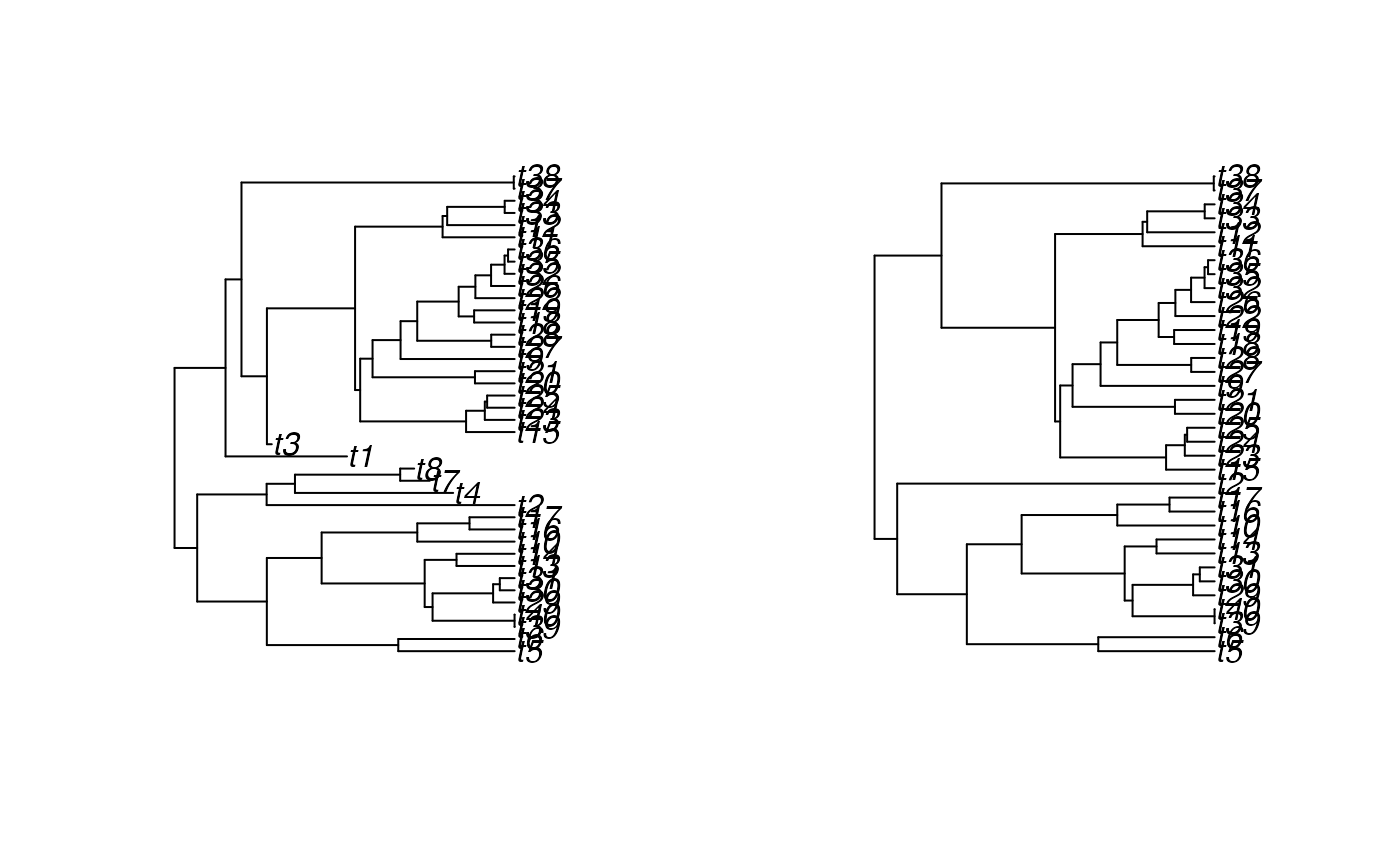
plot(SimToPhylo(simulated.result$results, include.extinct=TRUE))
plot(SimToPhylo(simulated.result$results, include.extinct=FALSE))
 # }
# }